
Reshma R. Rao, Manuel J. Kolb, Livia Giordano, Anders Filsøe Pedersen, Yu Katayama, Jonathan Hwang, Apurva Mehta, Hoydoo You, Jaclyn R. Lunger, Hua Zhou, Niels Bendtsen Halck, Tejs Vegge, Ib Chorkendorff, Ifan E. L. Stephens & Yang Shao-Horn
DOI: 10.1038/s41929-020‑0457‑6
Abstract:
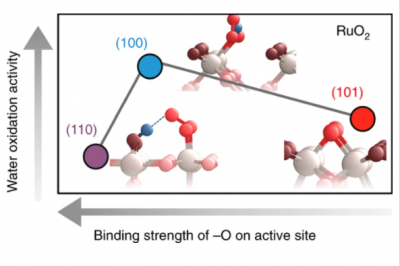
Understanding the nature of active sites is central to controlling (electro)catalytic activity. Here we employed surface X‑ray scattering coupled with density functional theory and surface-enhanced infrared absorption spectroscopy to examine the oxygen evolution reaction on RuO2 surfaces as a function of voltage. At 1.5 VRHE, our results suggest that there is an –OO group on the coordinatively unsaturated ruthenium (RuCUS) site of the (100) surface (and similarly for (110)), but adsorbed oxygen on the RuCUS site of (101). Density functional theory results indicate that the removal of –OO from the RuCUS site, which is stabilized by a hydrogen bond to a neighbouring –OH (–OO–H), could be the rate-determining step for (100) (similarly for (110)), where its reduced binding on (100) increased activity. A further reduction in binding energy on the RuCUS site of (101) resulted in a different rate-determining step (–O + H2O – (H+ + e−) → –OO–H) and decreased activity. Our study provides molecular details on the active sites, and the influence of their local coordination environment on activity.