Understanding the ligand activity in positive electrodes for Li-ion batteries
(Credit: Yang Yu)
My research interest lies in understanding the electronic structures of oxides through both experimental and computational tools and exploring how different electronic structures alter the redox process of the oxides and their interfacial reactivity. We started off taking one commonly used cathode family, Li nickel manganese cobalt oxides (NMC), as the model system, where we aim to understand the root cause for larger degree of capacity fade of cathodes with higher nickel contents, and the general degradation pathway of commercial carbonate-based electrolyte in contact with cathodes. We have shown through spectroscopy tools including Raman, Fourier-transform infrared spectroscopy (FT-IR) and X-ray photon electron spectroscopy (XPS), coupled with DFT calculations, that carbonate solvent tends to dissociative adsorb on the oxide surface by forming C-Olattice bond, creating surface protic species. This protic species will further decompose the salt, shown by FT-IR, XPS. Since carbonate decompose by forming bonds with lattice oxygen in the oxide upon decomposition, the energetics of this reaction is highly dependent on the O p-band center relative to the Fermi level, which explains that Ni-rich cathodes tends to have higher reactivity towards electrolyte due to high Ni-O covalency and closer O p-band towards Fermi level.1
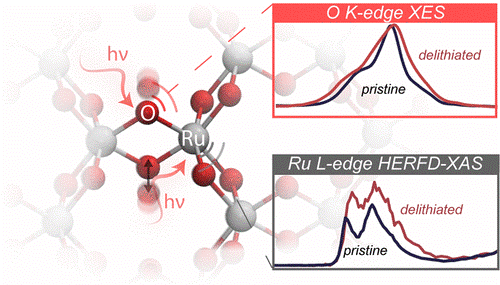
This ligand activity can be activated to the fullest when we consider the highly covalent metal oxides, for example, Li2RuO3, where the Fermi level is pinned on top of O p band. In the previous literatures, due to high covalency, Li2RuO3 are claimed to trigger peroxo-like species upon charging, where oxygen contributes partially to the capacity, yet there is no solid experimental evidence for such species. In our recent work, we have combined excited states DFT calculations as well as experimental X-ray absorption an emission spectra on the charged Li2RuO3, we can unequivocally pinpoint the electronic fingerprint of the anionic redox activities for the first time. In this work we have also highlighted the importance of covalency between transition metal and oxygen in keeping the oxygen framework stable by comparing with its 3d counterpart Li2MnO3, where less Mn-O leads to oxygen evolution.2
References
1. Yang, Y., Karayaylali, P., Katayama, Y., Giordano, L., Gauthier, L., Maglia, F., Jung, R., Lund, I. & Shao-Horn., Y. Coupled LiPF6 decomposition and carbonate dehydrogenation enhanced by highly covalent metal oxides in high-energy li-ion batteries. J. Phys. Chem. C 122 (48), 27368-27382 (2018).
2. Yang, Y., Karayaylali, P., Nowak, S. H., Giordano, L., Gauthier, M., Hong, W., Kou R., Li, Q., Vinson, J., Kroll, T., Sokaras, D., Sun, C. -J., Charles, N., Magliah, F. Jung, R., Shao-Horn, Y., Revealing electronic signatures of lattice oxygen redox in lithium ruthenates and implications for high-energy Li-ion battery material designs. Chem. Mat. 31 (19) 7864-7876 (2019).
In-Situ Operando Characterization of SEI for Lithium Metal Anodes
(Credit: Chris Mallia)
The safe and reliable implementation of lithium metal anodes has been a grail-like quest for researchers in the battery community for nearly fifty years.1,2 The energetic benefits are a high specific capacity (3860 mAh/g) and low mass density, as well as the lowest electrochemical potential of any reactive metal enabling higher power delivery. 3 With these benefits, lithium metal anodes are obvious choice to expand secondary and primary battery capacity to new limits. The problem confronting active use of lithium metal anodes largely remains in the poor cycling efficiency, plating kinetics, and the numerous parasitic reactions that occur with conventional battery electrolyte systems. 4,5 Nearly all known solvents react with lithium metal, forming what is known as the Solid Electrolyte Interphase (SEI) on the surface, which as its nomenclature describes, acts as an ion-conducting and electronically insulating solid electrolyte that prevents further reaction with lithium metal.6
Despite what would appear to be a stabilizing process, the formation of SEI on the surface of lithium metal has failed to adequately protect lithium metal in long-term battery cycling and even against open circuit discharge.7 SEI layers are often structurally heterogenous, kinetically unstable during the lithium plating process and as such continually breakdown and reform over charge/discharge cycles, a process which depletes active lithium ions in the electrolyte and “dries” out electrolyte solvents.8 In order to combat these destructive side effects of lithium metal, critical information about SEI chemical composition, dynamic behavior during cycling and ability to enable homogenous lithium plating is needed. To tackle this difficult problem, novel electrochemical techniques are necessary, and one non-destructive method is in-situ operando FTIR spectroscopy.9 Lithium metal anodes can be formed in-situ on the surface of an inert electrode, while real-time IR spectra of the anode surface can be taken. This technique allows for dynamic measurement of the composition of the SEI, while the cell is cycled under various conditions electrochemical conditions.
The knowledge gained by such characterization can enable choices for electrolyte chemistries that form adequately passivating layers on lithium metal. Likewise, ex-situ artificial SEI compositions can be tested in real cells and have chemical changes tracked real-time. These chemistries must be able to prevent further reduction of electrolyte species, and furthermore accommodate the motion of lithium ions and the change in the lithium metal anode morphology over many cycles. By combining operando and in-situ measurement techniques, SEI chemical composition can be correlated to bulk cell performance, and therefore indicate what chemical moieties are good candidates. These factors make it critical to understand the behavior of the SEI on a molecular level, and connect that information to the mesoscale performance of lithium metal anodes, so that better performance can be enabled.
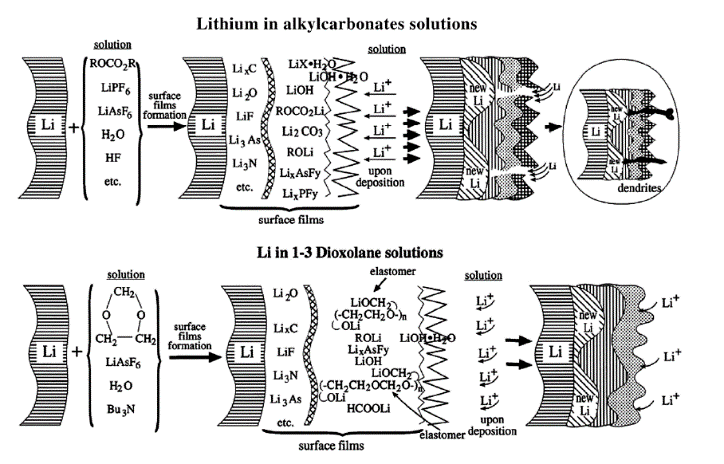
Figure 1. Schematic SEI formation in various non-aqueous battery systems. Reproduced from Ref. [8].
References:
1. Peled, E. The Electrochemical Behavior of Alkali and Alkaline Earth Metals in Nonaqueous Battery Systems—The Solid Electrolyte Interphase Model. J. Electrochem. Soc. 126 (12), 2047 (1979).
2. Xu, K. Nonaqueous Liquid Electrolytes for Lithium-Based Rechargeable Batteries. Chem. Rev. 104 (10), 4303–4418 (2004).
3. Xu, W., Wang, J., Ding, F., Chen, X., Nasybulin, E., Zhang, Y., Zhang, J.-G. Lithium Metal Anodes for Rechargeable Batteries. Energy Environ. Sci. 7 (2), 513–537 (2014).
4. Aurbach, D. A Short Review of Failure Mechanisms of Lithium Metal and Lithiated Graphite Anodes in Liquid Electrolyte Solutions. Solid State Ionics 148 (3–4), 405–416 (2002).
5. Aurbach, D., Markovsky, B., Levi, M. D., Levi, E., Schechter, A., Moshkovich, M., Cohen, Y. New Insights into the Interactions between Electrode Materials and Electrolyte Solutions for Advanced Nonaqueous Batteries. Journal of Power Sources 81–82, 95–111 (1999).
6. Wang, A., Kadam, S., Li, H., Shi, S., Qi, Y. Review on Modeling of the Anode Solid Electrolyte Interphase (SEI) for Lithium-Ion Batteries. npj Comput. Mater. 4 (1), 15 (2018).
7. Peled, E., Menkin, S. Review—SEI: Past, Present and Future. J. Electrochem. Soc. 164 (7), A1703–A1719 (2017).
8. Aurbach, D. Review of Selected Electrode–Solution Interactions Which Determine the Performance of Li and Li Ion Batteries. Journal of Power Sources 89 (2), 206–218 (2000).
9. Aurbach, D., Moshkovich, M., Cohen, Y., Schechter, A. The Study of Surface Film Formation on Noble-Metal Electrodes in Alkyl Carbonates/Li Salt Solutions, Using Simultaneous in Situ AFM, EQCM, FTIR, and EIS. Langmuir 15 (8), 2947–2960 (1999).